
Examples
Consider making a shortcut
See Getting Started to learn how to build Sequelizer. First, the build instructions leave you with a sequelizer binary in your build directory. You might want to alias it for easier use:
some_dir $ alias sequelizer='<whatever_your_path_is>/build/sequelizer'
Now you can run Sequelizer commands with sequelizer. The examples below assume you have done this.
Sequelizer Examines Fast5’s
You can use Sequelizer to analyze Fast5 files. A mature file format that holds nanopore information, both raw DNA measurements and lots of useful metadata. We (and you?) want circuits, computers, programs, networks working on this data to extract relevant genomic inisights anytime, anywhere.
To play around you’ll want some Fast5 files on your system. Here’s how to download a sample dataset
# Get some Fast5 files (this is ~2.4GB, with a 15MB/s download, it should take ~2.5 min)
some_dir $ wget https://ont-exd-int-s3-euwst1-epi2me-labs.s3-eu-west-1.amazonaws.com/fast5_tutorial/sample_fast5.tar
# Uncompress
some_dir $ tar -xvf sample_fast5.tar
# See what you got
some_dir $ ls
... sample_fast5/ ...
Now that you have some data in the sample_fast5 directory, you can use Sequelizer to analyze the Fast5 files. You do this using the package’s fast5 subcommand. Just point it to the directory with the --recursive flag to find all Fast5 files within:
# sequelize it
some_dir $ sequelizer fast5 sample_fast5 --recursive
Discovering Fast5 files...
Found 5 files, analyzing...
[[████████████████████████████████████████] 100% (5/5)
Sequelizer Fast5 Dataset Analysis Summary
=========================================
Files processed: 5/5 successful
Total file size: 2440.0 MB
Total reads: 20000
Signal statistics:
Total samples: 679508454
Average length: 33975 samples
Range: 1978 - 1199785 samples
Average bits per sample: 28.73
Total duration: 2831.3 minutes
Avg duration: 8.5 seconds
Processing time: 43.02 seconds
What happened? Sequelizer scanned the Fast5 files, extracted some basic statistics about the raw signal data, and printed a summary report. You can see how many reads were processed in the directory you scanned, their lengths, and other useful information. Almost 30 bits per sample were used to store the raw signal data? Awful if true.
If you want to know a little more about each read that was processed you could ask for a summary report to be made.
some_dir $ sequelizer fast5 sample_fast5 --recursive --summary
This will create a sequelizer_summary.txt file in your current directory with a line for each read processed, and some basic statistics about it. For example:
#sequelizer_summary_v1.0
filename read_id run_id channel start_time translocation_time num_samples median_before
FAK42335_2bf4f211a2e2d04662e50f27448cfd99dafbd7ee_0.fast5 00058fe1-e555-4a64-a41b-7f58fb7d6d6b 2bf4f211a2e2d04662e50f27448cfd99dafbd7ee 381 52.1 6.2 24688 204.58
FAK42335_2bf4f211a2e2d04662e50f27448cfd99dafbd7ee_0.fast5 000dd482-c0d5-4520-aa86-8ee8bb61fd58 2bf4f211a2e2d04662e50f27448cfd99dafbd7ee 323 12.3 6.6 26454 219.28
FAK42335_2bf4f211a2e2d04662e50f27448cfd99dafbd7ee_0.fast5 00158d74-4b7f-445a-b0ac-e1606f6c09b7 2bf4f211a2e2d04662e50f27448cfd99dafbd7ee 412 111.4 5.5 21941 162.95
FAK42335_2bf4f211a2e2d04662e50f27448cfd99dafbd7ee_0.fast5 004a0bd2-edcf-4c2c-89bc-009a232cdb6a 2bf4f211a2e2d04662e50f27448cfd99dafbd7ee 477 127.8 6.6 26345 202.09
...
See fast5 Command for more details.
Sequelizer Converts Fast5’s
Sequelizer can also convert Fast5 files to other formats (well one format, for now). For example, you can convert Fast5 files to txt using the convert subcommand:
some_dir $ sequelizer convert sample_fast5 --to raw -r
[████████████████████████████████████████] 100% (5/5)
In this case, the --to raw flag tells Sequelizer to convert the Fast5 files to raw text files. The -r flag tells it to search recursively through the sample_fast5 directory for Fast5 files. After running this command, you will find .txt files in some_dir. If you want them to go in a different directory, say some_dir/signals/ then also include the command-line argument -o signals.
For single-read Fast5 files the default file name takes the form read_ch<channel num>_rd<read num>.txt. For multi-read Fast5 files the default file name takes the form <original fast5 files name>_read_ch<channe _num>_rd<read num>.txt. In this example, the Fast5 files in sample_fast5 are multi-read files, so you should see files like:
some_dir $ ls
FAK42335_2bf4f211a2e2d04662e50f27448cfd99dafbd7ee_0_read_ch323_rd17.txt
FAK42335_2bf4f211a2e2d04662e50f27448cfd99dafbd7ee_0_read_ch381_rd59.txt
FAK42335_2bf4f211a2e2d04662e50f27448cfd99dafbd7ee_0_read_ch412_rd172.txt
FAK42335_2bf4f211a2e2d04662e50f27448cfd99dafbd7ee_100_read_ch108_rd3140.txt
...
By default, convert only extracts the first three reads from each Fast5 file that it finds. You can change this with the --all flag which tells Sequelizer to extract all reads from each Fast5 file.
What’s inside each .txt file? Behold:
# Channel: 198
# Offset: 6.000000
# Range: 1440.801147
# Digitisation: 8192.000000
# Conversion: signal_pA = (raw_signal + offset) * range / digitisation
# Sample Rate: 4000.0
# Duration: 3846
# Read ID: 00213bd6-0b7d-4e96-862f-160852db369a
#
sample_index raw_sample
0 441
1 465
2 484
3 455
...
The header contains metadata about the read, including channel number, offset, range, digitization, conversion formula, sample rate, and read ID. Below the header is a two-column table with the sample index and the corresponding raw signal value. See convert Command for more details.
Sequelizer Plots Signals
Sequelizer can also plot the raw signal data. Once you have a set of .txt files (see above) you can use the plot subcommand to create plots of the signals. For example:
some_dir $ sequelizer plot ./signals/FAK42335_2bf4f211a2e2d04662e50f27448cfd99dafbd7ee_100_read_ch198_rd2884.txt --title "RAW data"
Should produce a plot like this (if you were wondering what DNA looks like, is just a squiggly line):
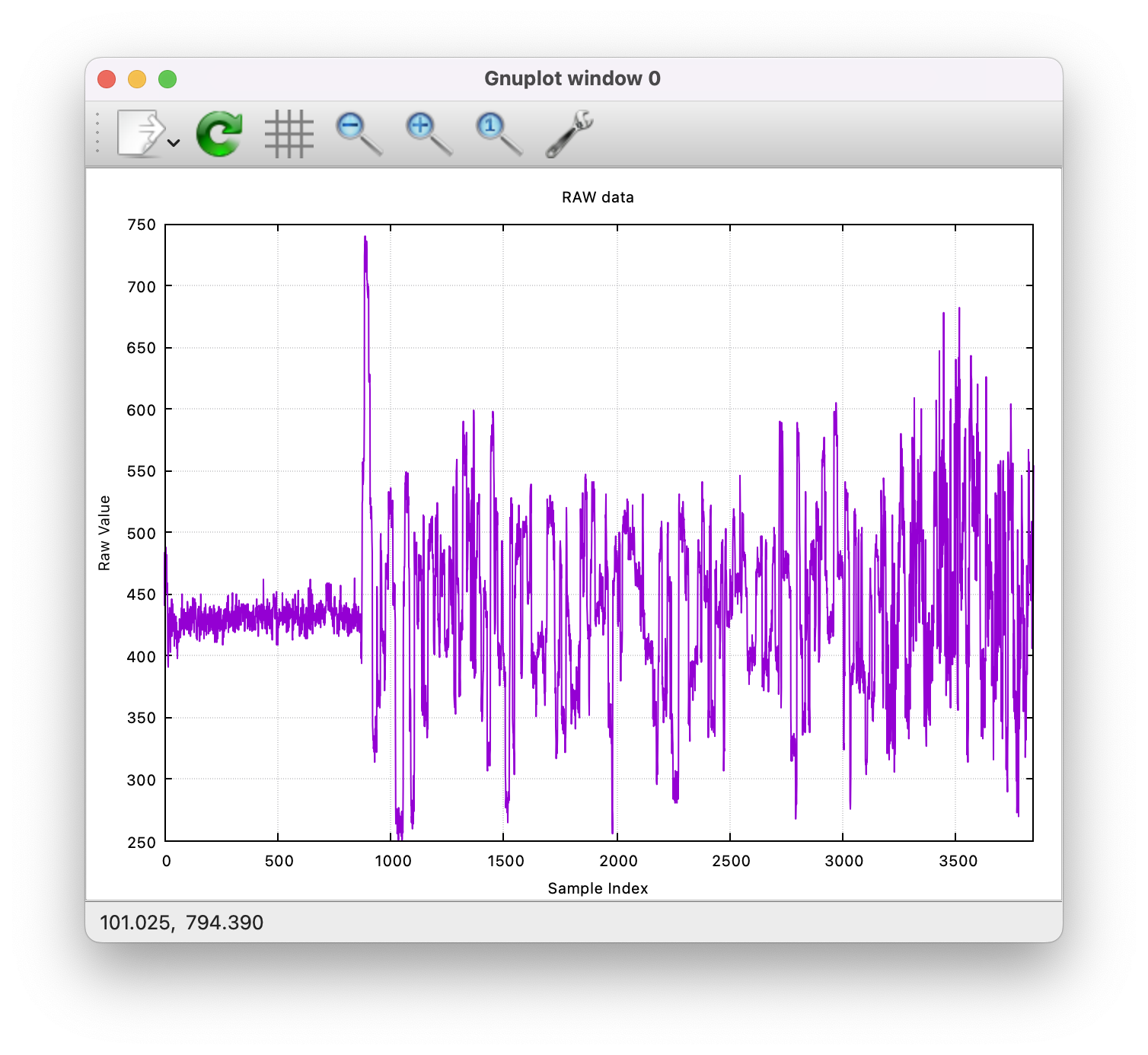
See plot Command for more details.
Sequelizer Makes Fake Signals
Sequelizer can also generate fake raw signal data from DNA sequences. This is useful for testing and development purposes. You can use the seqgen subcommand to create synthetic signals from DNA sequences in FASTA/FASTQ files or just generate random sequences yourself.
For example, in the sample_fast5 directory we downloaded you’ll find a bunch of FASTQ files (presumably generated from the raw data in the fast5 files located in sample_fast5/workspace). To generate signals from one of these FASTQ files, you can run:
some_dir $ sequelizer seqgen ./sample_fast5/fastq_runid_2bf4f211a2e2d04662e50f27448cfd99dafbd7ee_0_0.fastq --select 1 -o squiggle1.txt
seq length 2053
Average dwell time: 10.000000 (across 2049 positions and 20490 samples)
This command will create a synthetic signal for the first read (as directed by --select 1) in the specified FASTQ file and save it to squiggle1.txt. It’s reporting that it consumed 2053 letters (i.e., A’s, C’s, G’s, and T’s) and that it created a signal with 2049 signals that are distributed across 20490 samples (i.e., the samples that a nanopore’s analog-to-digital converter would take to produce such a signal). This is admittedly pretty artifical, if you explore you’ll find more realistic options. Here’s what the inside of that file should look like:
#6ea6609b-2962-4ac4-a792-5375fb9b2d23
pos base current sd dwell
0 C 1.482633 1.500000 10.000000
1 A 0.705864 1.500000 10.000000
2 G 0.093573 1.500000 10.000000
3 T 1.183438 1.500000 10.000000
4 G -0.591415 1.500000 10.000000
...
The file contains a header with the read ID, followed by a table with columns for position, base, current in pA (picoamperes), standard deviation, and dwell time (in terms of samples).
You can customize the output using various options provided by the seqgen command. See seqgen Command for more details.
Before You Go
Please help Sequelizer development. Tell us what else you’d like to see here!